Cystisk fibrose

Cystisk fibrose er en sykdom som får tykt, klebrig slim til å bygge seg opp i lungene, fordøyelseskanalen og andre områder av kroppen. Det er en av de vanligste kroniske lungesykdommene hos barn og unge voksne. Det er en livstruende lidelse.
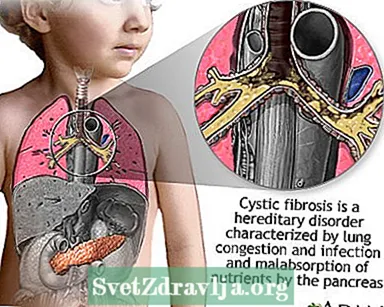
Cystisk fibrose (CF) er en sykdom som overføres gjennom familier. Det er forårsaket av et defekt gen som får kroppen til å produsere unormalt tykk og klebrig væske, kalt slim. Dette slimet bygger seg opp i lungene og i bukspyttkjertelen.
Oppbyggingen av slim resulterer i livstruende lungeinfeksjoner og alvorlige fordøyelsesproblemer. Sykdommen kan også påvirke svettekjertlene og reproduksjonssystemet til en mann.
Mange har et CF-gen, men har ikke symptomer. Dette er fordi en person med CF må arve 2 defekte gener, 1 fra hver av foreldrene. Noen amerikanere har CF-genet. Det er mer vanlig blant de av nord- eller sentraleuropeisk avstamning.
De fleste barn med CF får diagnosen 2 år, spesielt da screening av nyfødte utføres over hele USA. For et lite antall blir sykdommen ikke oppdaget før 18 år eller eldre. Disse barna har ofte en mildere form av sykdommen.
Symptomer hos nyfødte kan omfatte:
- Forsinket vekst
- Unnlatelse av å gå opp i vekt normalt i barndommen
- Ingen avføring i løpet av de første 24 til 48 timene av livet
- Salt smakende hud
Symptomer relatert til tarmfunksjon kan omfatte:
- Magesmerter fra alvorlig forstoppelse
- Økt gass, oppblåsthet eller mage som virker hovent (utspent)
- Kvalme og tap av matlyst
- Avføring som er blek eller leirefarget, illeluktende, har slim eller som flyter
- Vekttap
Symptomer relatert til lungene og bihulene kan omfatte:
- Hoste eller økt slim i bihulene eller lungene
- Utmattelse
- Nesetetthet forårsaket av nesepolypper
- Gjentatte episoder av lungebetennelse (symptomer på lungebetennelse hos noen med cystisk fibrose inkluderer feber, økt hoste og kortpustethet, økt slim og tap av appetitt)
- Sinus smerte eller trykk forårsaket av infeksjon eller polypper
Symptomer som kan bli lagt merke til senere i livet:
- Infertilitet (hos menn)
- Gjentatt betennelse i bukspyttkjertelen (pankreatitt)
- Åndedrettssymptomer
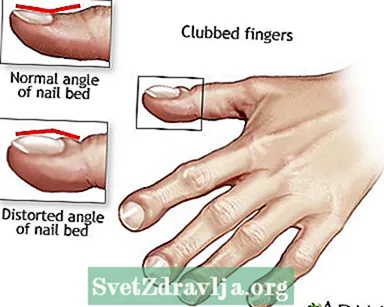
- Knebete fingre
En blodprøve gjøres for å oppdage CF. Testen ser etter endringer i CF-genet. Andre tester som brukes til å diagnostisere CF inkluderer:
- Immunoreaktiv trypsinogen (IRT) test er en standard nyfødt screening test for CF. Et høyt nivå av IRT antyder mulig CF og krever ytterligere testing.
- Svettekloridtest er standard diagnostisk test for CF. Et høyt saltnivå i svetten til personen er et tegn på sykdommen.
Andre tester som identifiserer problemer som kan relateres til CF inkluderer:
- Røntgen- eller CT-skanning på brystet
- Fekal fettprøve
- Lungefunksjonstester
- Måling av bukspyttkjertelfunksjon (avføring pankreas elastase)
- Secretin stimulering test
- Trypsin og chymotrypsin i avføring
- Øvre GI og tynntarmserier
- Lungekulturer (oppnådd ved sputum, bronkoskopi eller svelle)
En tidlig diagnose av CF og behandlingsplan kan forbedre både overlevelse og livskvalitet. Oppfølging og overvåking er veldig viktig. Når det er mulig, bør du ta vare på en spesialklinikk for cystisk fibrose. Når barn blir voksen, bør de overføre til et spesialistsenter for cystisk fibrose for voksne.
Behandling for lungeproblemer inkluderer:
- Antibiotika for å forebygge og behandle lunge- og bihuleinfeksjoner. De kan tas gjennom munnen, eller gis i venene eller ved pustebehandlinger. Personer med CF kan ta antibiotika bare når det er nødvendig, eller hele tiden. Doser er ofte høyere enn normalt.
- Inhalerte medisiner for å åpne luftveiene.
- Andre medisiner som gis ved pustebehandling for tynn slim og som gjør det lettere å hoste opp, er DNA-enzymbehandling og høykonsentrerte saltløsninger (hyperton saltoppløsning).
- Influensavaksine og pneumokokk polysakkaridvaksine (PPV) årlig (spør helsepersonell).
- Lungetransplantasjon er et alternativ i noen tilfeller.
- Oksygenbehandling kan være nødvendig når lungesykdommen blir verre.
Lungeproblemer behandles også med terapier for å tynne slimet. Dette gjør det lettere å hoste slimet ut av lungene.
Disse metodene inkluderer:
- Aktivitet eller trening som får deg til å puste dypt
- Enheter som brukes på dagtid for å fjerne luftveiene for mye slim
- Manuell perkusjon av brystet (eller fysioterapi på brystet), der et familiemedlem eller en terapeut lett klapper personens bryst, rygg og område under armene
Behandling for tarm- og ernæringsproblemer kan omfatte:
- Et spesielt kosthold med høyt proteininnhold og kalorier for eldre barn og voksne
- Bukspyttkjertelenzymer som hjelper til med å absorbere fett og protein, som tas med hvert måltid
- Vitamintilskudd, spesielt vitamin A, D, E og K
- Din leverandør kan gi råd om andre behandlinger hvis du har veldig harde avføring
Ivacaftor, lumacaftor, tezacaftor og elexacaftor er medisiner som behandler visse typer CF.
- De forbedrer funksjonen til et av de defekte genene som forårsaker CF.
- Opptil 90% av pasientene med CF og er berettiget til en eller flere av disse medisinene alene eller i kombinasjon.
- Som et resultat er det mindre opphopning av tykt slim i lungene. Andre CF-symptomer forbedres også.
Omsorg og overvåking hjemme bør omfatte:
- Unngå røyk, støv, smuss, røyk, kjemikalier til husholdningene, peisrøyk og mugg eller mugg.
- Å gi rikelig med væske, spesielt til spedbarn og barn i varmt vær, når det er diaré eller løs avføring, eller under ekstra fysisk aktivitet.
- Trener 2 eller 3 ganger hver uke. Svømming, jogging og sykling er gode alternativer.
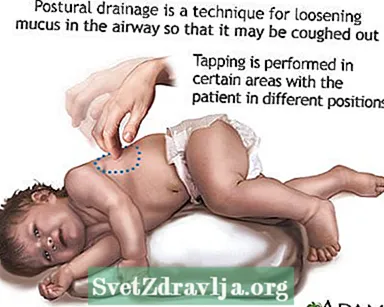
- Å fjerne eller bringe opp slim eller sekreter fra luftveiene. Dette må gjøres 1 til 4 ganger hver dag. Pasienter, familier og omsorgspersoner må lære om å gjøre perkusjon av brystet og postural drenering for å holde luftveiene klare.
- Ingen kontakt med andre personer med CF anbefales fordi de kan utveksle infeksjoner (gjelder ikke familiemedlemmer).
Du kan lette stresset ved sykdom ved å bli med i en støttegruppe for cystisk fibrose. Å dele med andre som har vanlige erfaringer og problemer kan hjelpe familien din til ikke å føle seg alene.
De fleste barn med CF har god helse til de blir voksne. De er i stand til å delta i de fleste aktiviteter og gå på skole. Mange unge voksne med CF fullfører college eller finner seg jobber.
Lungesykdom forverres til slutt til det punktet hvor personen er ufør. I dag er den gjennomsnittlige levetiden for personer med CF som lever til voksen alder ca 44 år.
Døden er oftest forårsaket av lungekomplikasjoner.
Den vanligste komplikasjonen er kronisk luftveisinfeksjon.
Andre komplikasjoner inkluderer:
- Tarmproblemer, som gallestein, tarmblokkering og endetarmsutfall
- Hoste opp blod
- Kronisk respirasjonssvikt
- Diabetes
- Infertilitet
- Leversykdom eller leversvikt, pankreatitt, galle cirrhose
- Underernæring
- Nesepolypper og bihulebetennelse
- Osteoporose og leddgikt
- Lungebetennelse som stadig kommer tilbake
- Pneumothorax
- Høyresidig hjertesvikt (cor pulmonale)
- Tykktarmskreft
Ring leverandøren din hvis et spedbarn eller barn har symptomer på CF, og opplever:
- Feber, økt hoste, endringer i sputum eller blod i sputum, tap av appetitt eller andre tegn på lungebetennelse
- Økt vekttap
- Hyppigere avføring eller avføring som lukter vondt eller har mer slim
- Hovent mage eller økt oppblåsthet
Ring leverandøren din hvis en person med CF utvikler nye symptomer eller hvis symptomene blir verre, spesielt alvorlige pustevansker eller hoste opp blod.
CF kan ikke forhindres. Screening av de med en familiehistorie av sykdommen kan oppdage CF-genet i mange bærere.
CF
- Enteral ernæring - barn - håndterer problemer
- Gastrostomi fôringsrør - bolus
- Hvordan puste når du er kortpustet
- Jejunostomy fôringsrør
- Postural drenering
 Klubb
Klubb Postural drenering
Postural drenering Knebete fingre
Knebete fingre Cystisk fibrose
Cystisk fibrose
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor / ivacaftor hos personer med cystisk fibrose og F508del / F508del-CFTR eller F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Cystisk fibrose. I: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, red. Nelson lærebok for pediatri. 21. utg. Philadelphia, PA: Elsevier; 2020: kap 432.
Farrell PM, White TB, Ren CL, et al. Diagnose av cystisk fibrose: konsensusretningslinjer fra Cystic Fibrosis Foundation. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effekter av lumacaftor / ivacaftor-terapi på CFTR-funksjon hos Phe508del homozygote pasienter med cystisk fibrose. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Cystisk fibrose. I: Goldman L, Schafer AI, red. Goldman-Cecil Medicine. 26. utg. Philadelphia, PA: Elsevier; 2020: kap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Cystisk fibrose. I: Broaddus VC, Mason RJ, Ernst JD, et al, red. Murray og Nadels lærebok for respiratorisk medisin. 6. utg. Philadelphia, PA: Elsevier Saunders; 2016: kap 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor hos pasienter med cystisk fibrose homozygot for phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Interessante Publikasjoner

Michelle Obama lanserer en podcast for å styrke forholdet ditt til andre – og deg selv